A beginner’s guide to imaging in vivo tumour biochemistry
Tim Witney
Here we discuss how imaging can be used to understand biochemical processes in living subjects without the need for invasive procedures. It’s written for a non-specialist audience and is a good starting point to learn more about the subject.
The article was originally published in The Biochemist and is authored by Hannah Greenwood, Richard Edwards and Tim Witney from the Molecular Imaging Lab.
Using antimatter to look at cancer
Cancer can be defined as the uncontrolled proliferation of cells in the body that arise from genetic mutation. Cancer is a devastating disease, accounting for more than a quarter of all deaths in the UK (Cancer Research UK, https://www.cancerresearchuk.org/health-professional/cancer-statistics-for-the-uk, accessed May 2019). Over the last couple of decades, a detailed understanding of the molecular pathways associated with cancer growth, progression and metastases has led to the development of new and improved treatments that target these pathways. For example, the identification of the HER2 receptor, with the resulting drug, Herceptin, has produced long-term survival benefits in many women with HER2-positive breast cancer. We are able to characterise, measure and visualise these important biochemical processes in a living system using a powerful medical imaging technique called positron emission tomography (PET). Unlike other medical imaging technologies, such as computed tomography or magnetic resonance imaging, which provide detailed anatomical information, PET can reveal the functional processes that drive disease without the need for surgical intervention. In this article, we will discuss the steps involved in developing ‘smart’ imaging probes to visualise in vivo tumour biochemistry with PET.
Cancer is a heterogenous disease, with multiple phenotypes observed both between different metastatic sites and within the primary tumour itself. Tumour heterogeneity has wide-spread implications for patient diagnosis, treatment selection, and ultimately how these tumours respond to therapy. Diagnosis is traditionally confirmed by biopsy following referral by a GP, often after the detection of a suspicious growth or the onset of symptoms. Biopsies are an invasive procedure in which a small region of the suspected lesion is surgically excised and examined under a microscope to assess the presence (or absence) of malignant cells. Tumour biopsies, however, hold a number of limitations: As only a small region of the tumour is sampled, a single biopsy may not capture the whole picture of the disease itself. In extreme cases, inadequate sampling can lead to the misdiagnosis and wrongful treatment of patients. In addition, acquiring tumour biopsies is an invasive procedure, and in some cases a biopsy cannot be obtained at all due to the location of the mass. By comparison, PET imaging can provide doctors with whole-body images of cancer biochemistry. This allows the assessment of both primary tumours and distant metastases. Seeing ‘the whole picture’ allows for more accurate diagnosis and improved patient care.
PET imaging works through the indirect detection of an antimatter particle called a positron, emitted from an unstable atom, known as a radionuclide. These radioactive atoms can be attached to a molecule of biological interest to produce a radiolabelled molecule known as a radiotracer. Following its intravenous injection into a subject, the radiotracer accumulates at the selected biological target of interest and is detected by a PET scanner. A detailed description of this process is described in Figure 1.

Figure 1. Preclinical PET imaging. Prior to acquiring a PET image, a radiotracer is injected into the subject (1). Radiotracers are made by attaching a radioactive atom (radionuclide) to a scaffold that targets a biological process of interest. As the unstable radioactive atom decays it emits a positron (the antimatter equivalent of an electron), which travels a shorts distance (~1 mm) before colliding with a near-by electron (2). Upon collision, these two particles undergo a process called ‘annihilation’, releasing two gamma rays at ~180 degrees to one another. The detection of these gamma rays by a PET scanner allows for the origin of annihilation to be calculated, subsequently providing the precise location of the radiotracer. Computer processing enables multiple annihilation events to be processed, building a 3D ‘map’ of radioactivity inside the body – the PET image (3).
Designing smart imaging probes
If properly designed, a radiotracer can allow us to visualise important molecular processes relating to cancer. Before designing the radiotracer itself, an informative biological target to image must be selected. This biological target is called a ‘biomarker’. A biomarker can be an indicator of normal biological processes, disease processes or responses to an intervention, such as drug treatment. Important examples of biomarkers relating to PET imaging include receptors, enzymes, proteins, and transporters. The data obtained by the measurement of a well-selected biomarker can then be used to answer important clinical or fundamental question relating to cancer biology.
In a clinical setting, imaging will look to address a question regarding how to treat a patient. This could be ‘Does this patient have cancer?’, ‘How is the patient’s cancer responding to their therapy?’, ‘Will the patient’s cancer respond to this type of therapy?’ or even ‘What is the cancer phenotype?’. The different biomarkers exhibited by cancer cells can be targeted by PET probes to achieve selective uptake and accumulation in specific cancer types to answer these fundamental questions. This accurate characterisation of the cancer allows for treatments to be selected for individual patients rather than the population as a whole in a process known as precision medicine.
Successful visualisation of the biomarker relies on ‘contrast’. Firstly, this means that the target must be differentially expressed in cancer over that of the background tissue, with any background uptake of the radiotracer clearing within the lifetime of the radioactive atom. Without this clearance, contrast cannot be achieved and visualisation of this biomarker will be not be possible. Finally, for the visualisation to be informative, the radiotracer must be delivered to, and specifically interact with its biological target. This requires appropriate design and application of the radiotracer.
An excellent example of a PET radiotracer for cancer imaging is [18F]FDG, a glucose analogue which is taken into cells by glucose transporters and phosphorylated by the enzyme hexokinase (Figure 2A). The replacement of a hydroxyl group for the radioactive fluorine isotope means that after intracellular phosphorylation the sugar becomes trapped as due to its altered structure it cannot be further metabolised. The increased glucose metabolism exhibited by cancer cells means that [18F]FDG is preferentially taken up, allowing for high contrast imaging of tumours. This is a perfect example of how a radiotracer can be used to measure altered cancer biochemistry, which may inform treatment decisions. [18F]FDG is now used in most modern hospitals for disease staging and confirmation of primary diagnoses. As we move towards more personalised medicine regimens, however, there is a requirement for probes not only to detect cancer but to distinguish cancers of a specific phenotype or aggressiveness. This requires the development of new, ‘smart’ imaging probes. In the design of these informative probes, a number of considerations must be made:
1. Where is the biological process occurring?
In order to image a biochemical process, the probe needs to be able to interact with the system under investigation. Delivering molecules and biological scaffolds to where you want them in the body is far from trivial. It is a problem that drug companies and their medicinal chemistry teams spend billions of pounds trying to solve. The same problems need to be addressed in PET imaging. Firstly, the radiotracer needs to get to the site of disease. This can be tricky as the complex in vivo environment means that many compounds are quickly cleared (removed from circulation) or broken down in the body. The compound may also need to cross physical barriers such as the blood brain barrier or a cell membrane. Correct selection and/or design of the radioactive probe is key to overcome these issues. As a result, the design of the radiotracer will vary considerably depending on the location of your biomarker, not only within the body, but at the cellular level. The properties of a radiotracer designed for an extracellular target can vary dramatically from a radiotracer imaging an intracellular or transmembrane target, as will be discussed below.
2. What type of scaffold will I use?
Scaffolds that can be radiolabelled for the production of PET radiotracers include small molecules, peptides, proteins and antibodies. Each of these scaffolds can target biomarkers to produce an informative image.
Small molecules, such as inhibitors or substrates can target intracellular proteins or enzymes. The intracellular location of the target means that the radiolabelled probe must cross the cell membrane. This may require the design and tuning of a small molecule’s properties, whilst radiolabelled endogenous species, such as sugars and amino acids, may have mechanisms for cell internalisation, often via membrane transporters. The probe can accumulate in the cell by binding to its recognition site (Figure 2B). Alternatively, a substrate-based probe can undergo a modification that leads to its ‘trapping’ in the cell. This ‘trapping’ can be due to alterations leading to an inability to cross the cell membrane once it enters the cell (e.g. phosphorylation of FDG - shown in Figure 2A) or binding at another intracellular site. Small molecule cell transporter substrates can also be radiolabelled to detect differences in cell transporter expression.
Peptides, proteins and antibodies are typically used to detect extracellular biomarkers. Advantageously, any issues regarding the cell membrane permeability of these scaffolds are negated for extracellular biomarkers. Radiolabelled versions of these scaffolds bind to specific receptors on the cell surface. This allows visualisation of tumours exhibiting increased receptor expression compared to healthy cells (Figure 2C). This can inform a clinician if a patient is likely to respond to therapy targeting this particular cancer type, e.g. HER2-expressing tumours.
![Figure 2. Different radiolabelled scaffolds and their applications. A) [18F]FDG is transported in to cells where it accumulates due to metabolic trapping subsequent to phosphorylation by hexokinase. B) A radiolabelled drug molecule crosses the cellu…](https://images.squarespace-cdn.com/content/v1/53db178ce4b07b7551af4b99/1565002879783-O87DX5OS8O4C0M31UQZ3/Figure+2.PNG)
Figure 2. Different radiolabelled scaffolds and their applications. A) [18F]FDG is transported in to cells where it accumulates due to metabolic trapping subsequent to phosphorylation by hexokinase. B) A radiolabelled drug molecule crosses the cellular membrane and binds to its intracellular protein target. C) Radiolabelled antibodies bind to the extracellular domain of specific receptors.
Synthesis of smart imaging probes
The synthesis of radioactive molecules (termed radiochemistry) has unique challenges, most prevalent of which is the synthesis and purification of the radiotracer within the time constraints of the decaying radionuclide. Production and purification should normally be completed within 2 half-lives of the radioactive atom (before 75% of the starting radioactivity has decayed) so there is enough radioactive material left to enable reliable imaging of the disease. Synthesis is further complicated when using large amounts of radioactivity. While the opening credits of ‘The Simpsons’ may suggest that a biohazard suit and a pair of tongs are suitable protection for handling radioactive material, this certainly isn’t the case for PET radionuclides! The gamma radiation produced is highly penetrating and is what permits the deep tissue visualisation of PET probes. This means that standard organic chemistry procedures are impossible, or certainly not advisable! To overcome these problems, robotic systems have been developed to perform the desired manipulations in an automated fashion. These systems can then be placed in a heavily shielded compartment known as a ‘hot cell’ and controlled remotely.
The radiolabelling process depends on the scaffold chosen and the radioactive atom (radionuclide) selected. The half-life of your radionuclide should match that of the biological process you’re interested in visualising – you don’t want all of your radiotracer to decay before the meaningful data is acquired. For example, the radionuclide fluorine-18 (half-life of about 2 hours) is very popular for labelling small drug molecules, as these compounds are rapidly removed from the blood and excreted. This allows contrast, and therefore visualisation, of your target a short time after injection. Labelling an antibody with fluorine-18 would be impractical, however, as the radiotracer would completely decay before its removal from the blood, which can take many days. For visualisation of a labelled antibody’s target, better contrast is achieved much later. Therefore, a radionuclide with a longer half-life is generally used, such as zirconium-89 (half-life of about 3 days), so that contrast can be obtained. Once the radionuclide and the scaffold being labelled have been selected, a number of strategies are available to incorporate or attach the radioactive atom.
One approach is to produce a direct radioactive equivalent of your molecule of interest (Figure 3A). Advances in both radiochemistry and the robotic platforms used to perform the manipulations, have led to remarkable advances in our ability to incorporate radioactive atoms into structures of increasingly complexity. The radiolabelling can be performed using the radionuclide itself (e.g. fluorine-18 fluoride for fluorination) or by using radioactive reagents or building blocks (e.g. carbon-11 methyliodide for methylation) to synthesise the molecule. Alternatively, small molecules or peptides can have small radioactive units known as ‘prosthetic groups’ attached to them to facilitate radiolabelling where direct substitution of a non-radioactive atom for one that’s radioactive is not possible (Figure 3B). Larger species such as antibodies are amenable to more considerable alteration, whilst maintaining their original biological behaviour. This allows for functionalisation with larger structures such as chelators for coordinating radioactive metals including zirconium-89 or copper-64 (Figure 3C).
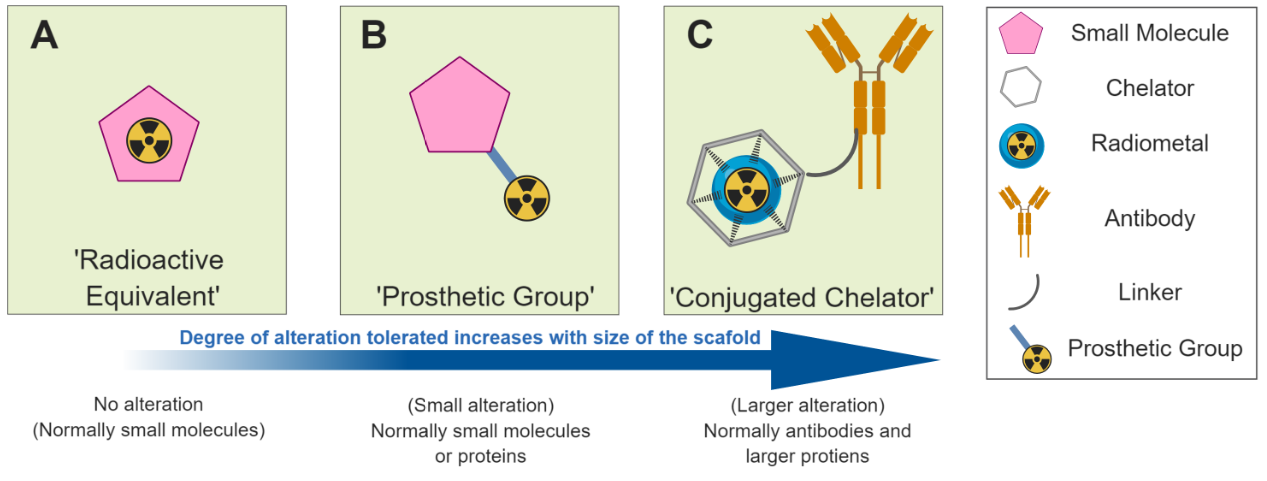
Figure 3. Different strategies for radiolabelling
As our understanding of how to deliver a radiotracer to its desired target progresses, radiotracer design can improve. In most cases, success will not be achieved at the first attempt. Novel radiotracers must be validated biologically (as discussed below). If the in vitro or in vivo studies show that the compound is not fulfilling the requirements of an informative radiotracer, the design and synthesis processes must start again using the new information acquired.
From radiosynthesis to imaging
Following the identification of an appropriate biomarker and development of the complementary imaging agent, cell-based validation must be completed to confirm the specificity of a probe to its target. Here, immortal cancer cell lines are incubated with the radioactive probe over a defined period of time. Scientists can then compare radiotracer accumulation or binding to cell lines with altered characteristics, for example, a drug sensitive vs. a drug resistant cell line, or cells expressing HER2 and those that do not. To check the selectivity of a probe to its target, blocking assays, competition assays or gene knock-down models are often used. Blocking and competition assays are used to prevent target-probe interactions. If the probe is specific to the biochemical target that has been blocked, no radioactivity should be detected in the cell. Knock-down models work in a similar way. Rather than blocking the target, the target gene and therefore biomarker is removed completely. If the novel probe is specific for the gene that has been knocked-out, there should be negligible radiotracer binding – demonstrating selectivity of the probe to the specific biomarker of interest. These initial experiments are necessary before the probe can be translated into complex living systems. Redesign of the imaging probe is required if non-specific binding is observed in these relatively simple models.
If the new radiotracer is shown to specifically interact with the biomarker of interest in cell culture-based experiments, validation then progresses to small animal research and in vivo (live animal) imaging. Tracking of the probe in healthy rodent models is performed prior to disease models. Experiments in healthy mice are important to understand the healthy tissue clearance and subsequent excretion route of the novel radiotracer. The two main excretion routes are via the renal system (kidneys and urine) and hepatobiliary system (liver and the intestines). The excretion route of the probe will determine which cancer types can be imaged. For example, a radiotracer that is cleared through the hepatobiliary system would not be appropriate to examine liver cancer due the high background signal in healthy liver tissue.
A crucial part of radiotracer assessment in vivo is the selection of clinically-relevant cancer models. The appropriate model could be a subcutaneously implanted patient-derived tumour xenograft using tissue taken directly from a cancer patient; an orthotopic tumour model, where tumour cell lines are grown in the tissue from which they originate; or a spontaneously-arising tumour generated in a genetically modified mouse, to name a few. Preclinical research provides vital information regarding a radiotracer’s binding to the target of interest, as well as information on how it is broken down. Dedicated small animal PET imaging scanners offer a platform for whole body, dynamic evaluation of these probes, often combined with computed tomography (CT) for anatomical reference. If the probe has been successfully delivered to its target, a ‘hotspot’ of radioactivity will be seen in the reconstructed PET image (Figure 4). The reconstructed PET image also allows scientists to observe the stability of the radiotracer. For example, when using a probe labelled with fluorine-18, if radioactive signal is seen in the bone, this is a sign of defluorination, where the radioisotope has become detached from its scaffold.
It is important to understand the in vivo binding properties, toxicity and radioactive dose delivery of novel imaging agents for Medicines and Healthcare products Regulatory Agency (MHRA) approval, prior to testing in humans. These tests are imperative for the complete validation of novel imaging agents but it must be noted that cell culture test or in vivo model will never replicate exactly what will be seen in humans. Clinical trials are critical in evaluating the efficacy of novel imaging agents in the diagnosis and management of diseases.

Figure 4. Preclinical versus human PET/CT imaging. Preclinical PET/CT allows whole-body visualisation of the radiotracer’s biodistribution, dynamically in small rodent models. In current clinical PET/CT systems, whole-body imaging is not possible as the whole body of the patient can not fit within the axial field of view of the scanner (usually 22 cm). This means only static scans, looking at the radiotracer distribution at a specific time, can be viewed. The clinical PET/CT image shown is a composite image from at least 2 or more bed positions. Developments in technology (and a lot of money) have led to the first total-body PET scanner – the EXPLORER.
The future of PET – antimatter matters
PET has advanced drastically from a technique used to simply visualise cancer lesions. One notable success is the emerging field of theranostics, which merges diagnostics and therapy into a single technique. As in conventional PET imaging, the first part of theranostic imaging is the diagnosis of a disease. A specially selected scaffold tagged with a PET radionuclide is injected into a patent where it will accumulate at the target of interest (if present). This accumulation of the radioactive probe will then be detecting during a PET scan. If there is high accumulation of the probe, the next part of theranostic imaging is to deliver a targeted amount of therapy to the patient. For this, the same scaffold is used; however, the gamma emitting radionuclide is replaced with a corresponding isotope that emits alpha particles. Alpha particles are highly charged and heavy atoms, and as a result they lose their energy in a very short distance, damaging the surrounding cells by causing double stand DNA breaks, for example. Specific delivery of these alpha particles to a cancerous lesion therefore gives a localised dose of therapy, which has been shown to be a highly effective treatment. This technique enables both selective imaging with the PET analogue, followed by therapy with the therapeutic equivalent. The most commonly used theronostic pair is technicium-99 and iodine-131 for the management of thyroid cancer. As well as thyroid cancer, theranostics has been extremely successful in the treatment of patients with neuroendocrine tumours and prostate cancer.
Excitingly, advances in scanner engineering have led to the development of the world’s first total-body PET scanner – the EXPLORER (Figure 4). The EXPLORER has an axial imaging field of view of 194 cm, almost 10-fold greater than a conventional PET/CT, allowing the entire body to be imaged at the same time. This milestone will provide previously unseen information on the whole-body dynamics of PET radiotracers. Lower doses, faster scanning and the ability to image at later time points will also be beneficial for patients and clinicians alike. The increased sensitivity that whole-body PET can offer will be particularly advantageous in cancer imaging, allowing the detection of small lesions that previously would go undetected.

Figure 5. The development of a novel PET imaging probe. Following the selection of a suitable biomarker, radiolabelling of the appropriate scaffold is performed. The radiotracer is first validated in cells to examine specificity and selectivity to the biomarker, before being injected into a mouse model for preclinical in vivo PET imaging. If the results of these tests show promising results, testing of the radiotracer moves into human clinical trials, following MHRA approval.
Conclusions
Imaging tumour biochemistry in vivo is a complex process that requires a collaborative approach from a multidisciplinary research team. We have discussed the processes that are involved in selecting a biochemical target to image, synthesising the probe and testing the radiotracer both in cells and in vivo prior to clinical translation (Figure 5). Non-invasive imaging of tumour biochemistry using new, smart imaging tools, places imaging scientists in a position to understand the mechanisms that drive cancer progression and the ability to treat the tumour whilst leaving healthy organs unaffected. The field of PET must strive to meet the demands of the medical community by delivering probes that can provide meaningful patient information, thereby improving the management and outcome of this devastating disease.
Further Reading
James, M. L. and Gambhir, S. S. (2012) A Molecular Imaging Primer: Modalities, Imaging Agents, and Applications. Physiol. Rev. 92, 897–965. doi: 10.1152/physrev.00049.2010 https://www.physiology.org/doi/full/10.1152/physrev.00049.2010
Witney, T. H. et al. (2015) PET imaging of tumor glycolysis downstream of hexokinase through noninvasive measurement of pyruvate kinase M2. Sci. Transl. Med. 7, 310ra169-310ra169. doi: 10.1126/scitranslmed.aac6117 https://stm.sciencemag.org/content/7/310/310ra169
Badawi, R. D. et al. (2019) First Human Imaging Studies with the EXPLORER Total-Body PET Scanner*. J. Nucl. Med. 60, 299–303. doi: 10.2967/jnumed.119.226498 http://jnm.snmjournals.org/content/60/3/299.long
van der Born, D. et al. (2017) Fluorine-18 labelled building blocks for PET tracer synthesis. Chem. Soc. Rev. 46, 4709–4773. doi: 10.1039/c6cs00492j https://pubs.rsc.org/en/content/articlehtml/2017/cs/c6cs00492j
Kostelnik, T. I. and Orvig, C. (2019) Radioactive Main Group and Rare Earth Metals for Imaging and Therapy. Chem. Rev. 119, 902–956. doi: 10.1021/acs.chemrev.8b00294 https://pubs.acs.org/doi/10.1021/acs.chemrev.8b00294